Viral Sequencing and analysis
Introduction
The pandemic lineage evolution in a short video nextclade
video “https://cardiff.cloud.panopto.eu/Panopto/Pages/Viewer.aspx?id=04f5593b-fe6e-4eb7-a69a-af3700e1e41e”
The derivation and viral lineages has played an essential part in our understanding and fight against the pandemic. The tutorial will take you through the steps where-by viral sequence is derived and analysed.
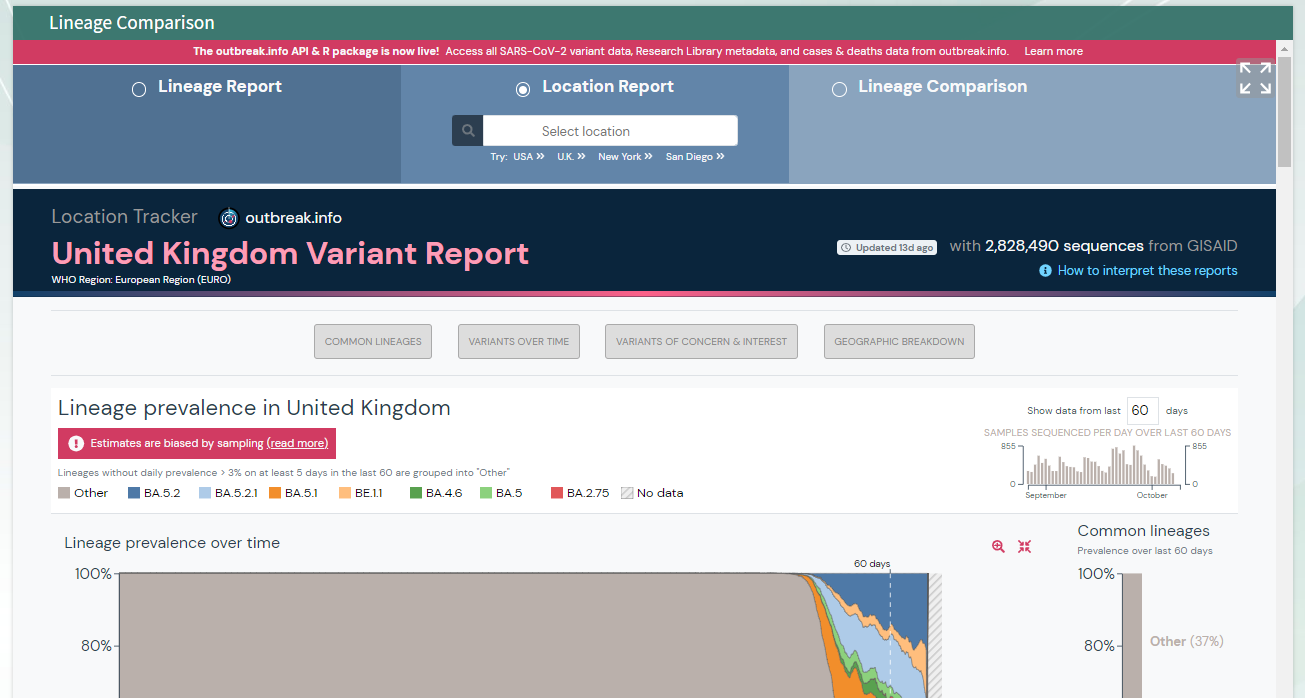
GISAID provides a range of resources to explore lineage prevalence and mutation comparison between lineages.
Here’s some key analyses it provides using the lineage sequence data:
Lineage Distribution (Omicron):
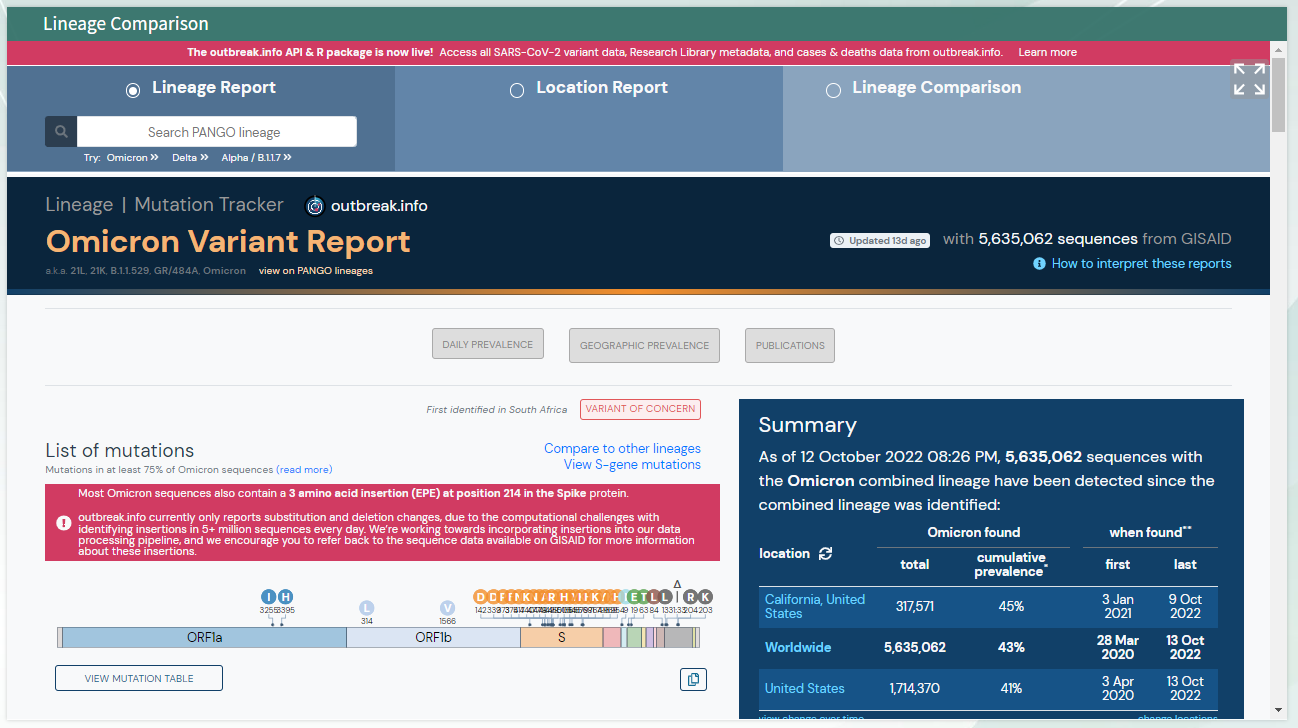
Lineage distribution Location in UK
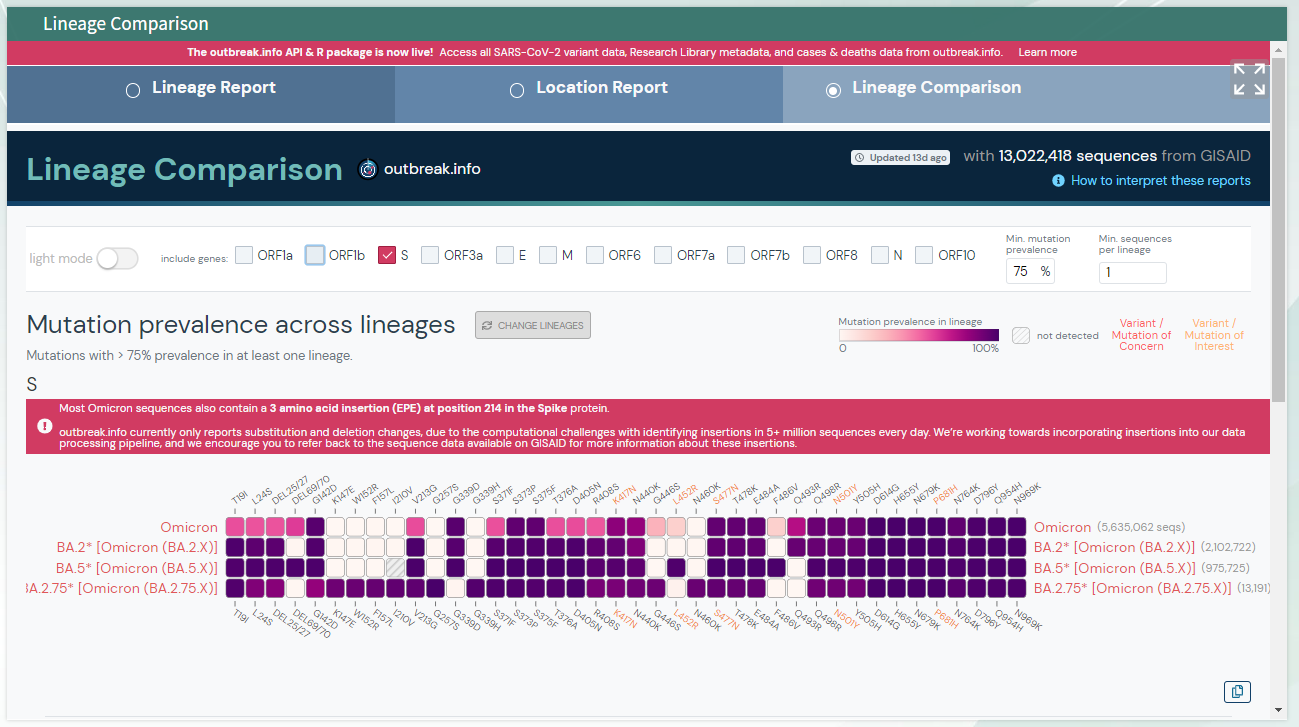
Mutation frequency across lineages
Viral sequencing
There a range of methods for deriving viral sequences from biological material. The basic steps are:
- Sample acquisition - saliva or Nasal swabs
- Nucleic acid extraction (RNA or DNA depending on virus)
- Reverse transcription (for RNA viruses)
- Viral enrichment (introduction of specimen specific indexes if multiplexing)
- Library constriction (introduction of specimen specific indexes if multiplexing)
- Sequencing (Illumina and Nanopore)
Steps 4 and 5 can be reversed if using capture enrichment approaches
Indexes can be introduced in steps 3 OR 4 but not both
A good review of these methods is provided by Gohl et al 2020 and is summarized in Figure 1. Also see Rosenthal et al., 2022.
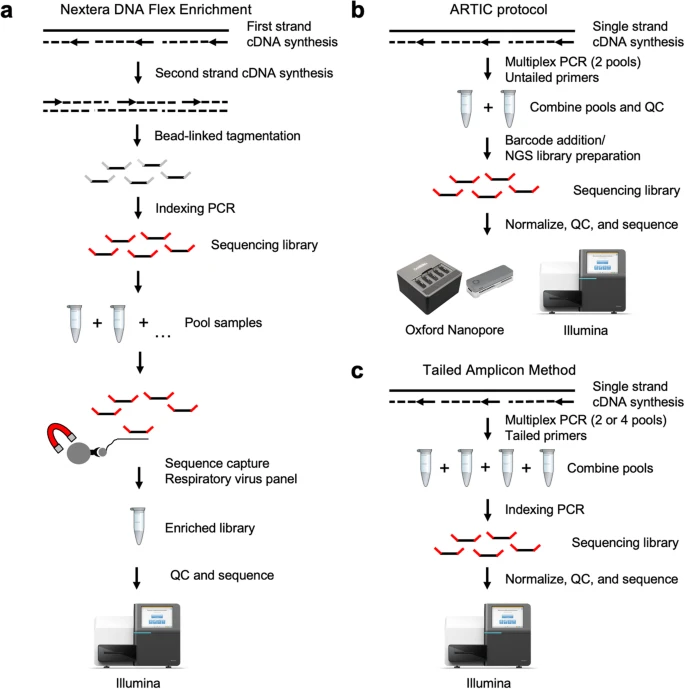
The approach that most used is what has been named the ARTIC protocol. This approach used 76 primer pairs to amplify the viral genome 2 sets of overlapping amplification reactions each containing 38 primer sets generating amplicons of ~400 bp. These amplicons were then pooled and sequenced (Illumina or Nanopore) to generate the complete genome.
Raw Data Sequence Analysis QC
- Quality assess and quality trim sequences
- Align sequences to reference covid genome (throw-away sequences that do not align)
- Evaluate coverage
- Mask primers used for amplification
- Derive consensus
- Determine Variants
Now some hands on data analysis, So lets do some analysis !!
Analysis Workflow of Raw Sequencing Data
Data
For the element of the workshop we have used Viral re-sequencing data generated using the ARTIC-V3 protocol from the COVID-19 Data Portal - EMBL. The following illumia paired end data was downloaded:
SRR13018901_1.fastq.gz SRR13635734_2.fastq.gz SRR15359995_1.fastq.gz SRR15360031_2.fastq.gz SRR15910995_1.fastq.gz SRR16192823_2.fastq.gz
SRR13018901_2.fastq.gz SRR13635763_1.fastq.gz SRR15359995_2.fastq.gz SRR15365583_1.fastq.gz SRR15910995_2.fastq.gz
SRR13635734_1.fastq.gz SRR13635763_2.fastq.gz SRR15360031_1.fastq.gz SRR15365583_2.fastq.gz SRR16192823_1.fastq.gzNote: We are using ‘pair end data’ some there is 2 sequences for each sample representing the forward and reverse samples.
Reference Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1 complete genome sequence (fasta) and annotation (gff) was downloaded from NCBI nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore/MN908947.3). (>send to > Complete Record > file > (FASTA/GFF3))
MN908947.3.fasta
MN908947.3.gffSoftware
cov2019 Artic Nextflow Workflow
Primary analysis work flow was performed using cov2019-artic-nf pipeline. This analysis was performed using a High Performance Linux (Ubuntu 20.04) Workstation (24 threads and 64 Gb RAM) running nexflow (https://www.nextflow.io/) and singularity (https://github.com/sylabs/singularity). command used to perform the analysis:
nextflow run connor-lab/ncov2019-artic-nf \ #analsyis pipline
-profile singularity \ #platform for software
--illumina \ #data type
--outdir $PWD/artic_analysis \ #output folder
--prefix "artic_test_run_221022" \ #output prefix
--directory $PWD/fastq_files/ #location of input files
--bed ~/covid_processing/artic_config_files/artic_V3.bed \ #primer sequences in bed format
--ref ~/covid_processing/nimagen_conf_files/MN908947.3.fasta \ #template sequence
--gff ~/covid_processing/nimagen_conf_files/MN908947.3.gff \ #template annotation
--yaml ~/covid_processing/nimagen_conf_files/SARS-CoV-2.types.yaml #covid classification fileYOU WILL NOT BE ABLE TO PERFORM THIS ANALYSIS, YOU WILL BE PROVIDED WITH THE OUTPUT OF THE PIPELINE WHICH CONTAINS THE FOLLOWING FOLDERS.
artic_analysis/
├── artic_test_run_221022.qc.csv
├── artic_test_run_221022.typing_summary.csv
├── artic_test_run_221022.variant_summary.csv
├── ncovIllumina_Genotyping_typeVariants
├── ncovIllumina_sequenceAnalysis_callVariants
├── ncovIllumina_sequenceAnalysis_makeConsensus
├── ncovIllumina_sequenceAnalysis_readMapping
├── ncovIllumina_sequenceAnalysis_readTrimming
├── ncovIllumina_sequenceAnalysis_trimPrimerSequences
├── qc_pass_climb_upload
└── qc_plotsGenome Visualisation Software - IGB
We will visualize the primary outputs using Integrated Genome Browser this can be downloaded and install on all platforms. It is also available as part of the BIOSI School software tools on the teaching workstation.
Workshop exercise
1. Review the QC summary and QC Plot
QC summary (this can be opened by opening artic_test_run_221022.qc.csv into excel)
| sample_name | pct_N_bases | pct_covered_bases | longest_no_N_run | num_aligned_reads | fasta | bam | qc_pass |
|---|---|---|---|---|---|---|---|
| SRR13018901 | 18.63 | 81.37 | 3133 | 492459 | SRR13018901.primertrimmed.consensus.fa | SRR13018901.mapped.primertrimmed.sorted.bam | TRUE |
Now review the linked QC plot that can be found in _analysis_plots
 Derive what the QC parameters are that would influence viral analsyis
Derive what the QC parameters are that would influence viral analsyis
2. Review and Overlay the reference genome (FASTA), annotation (GFF) and primer trimmed alignment file (bam)
Preview the fasta file and gff file using a text editor (wordpad / textpad on PC or Textedit on Mac). You cannot preview the bam file as it is a compressed binary file and is not human readable.
file locations:
Omics revolution course - Viral Genomics\Reference_files\MN908947.3.fasta
Omics revolution course - Viral Genomics\Reference_files\MN908947.3.gff
Omics revolution course - Viral\ Genomics\artic_analysis\ncovIllumina_sequenceAnalysis_trimPrimerSequences\SRR13018901.mapped.primertrimmed.sorted.bamFasta Preview (fasta format guide)
>MN908947.3 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAA
CGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACTCACGCAGTATAATTAATAAC
TAATTACTGTCGTTGACAGGACACGAGTAACTCGTCTATCTTCTGCAGGCTGCTTACGGTTTCGTCCGTG
.....GFF preview (GFF3 format guide)
##gff-version 3
##sequence-region MN908947.3 1 29903
#!genome-build ENA ASM985889v3
#!genome-version ASM985889v3
#!genome-date 2020-01
#!genome-build-accession NCBI:GCA_009858895.3
MN908947.3 ASM985889v3 region 1 29903 . . . ID=region:MN908947.3;Alias=NC_045512.2,NC_045512v2
####
MN908947.3 ensembl gene 266 13483 . + . ID=gene:ENSSASG00005000003;Name=ORF1ab;biotype=protein_coding;description=ORF1a polyprotein%3BORF1ab polyprotein [Source:NCBI gene (formerly Entrezgene)%3BAcc:43740578];gene_id=ENSSASG00005000003;logic_name=ensembl_covid;version=1
MN908947.3 ensembl mRNA 266 13483 . + . ID=transcript:ENSSAST00005000003;Parent=gene:ENSSASG00005000003;Name=ORF1a;biotype=protein_coding;transcript_id=ENSSAST00005000003;version=1
MN908947.3 ensembl exon 266 13483 . + . Parent=transcript:ENSSAST00005000003;Name=ENSSASE00005000003;constitutive=1;ensembl_end_phase=0;ensembl_phase=0;exon_id=ENSSASE00005000003;rank=1;version=1
....Now overlay these features using Integrated Genome Browser using the following steps:
- Open IGB
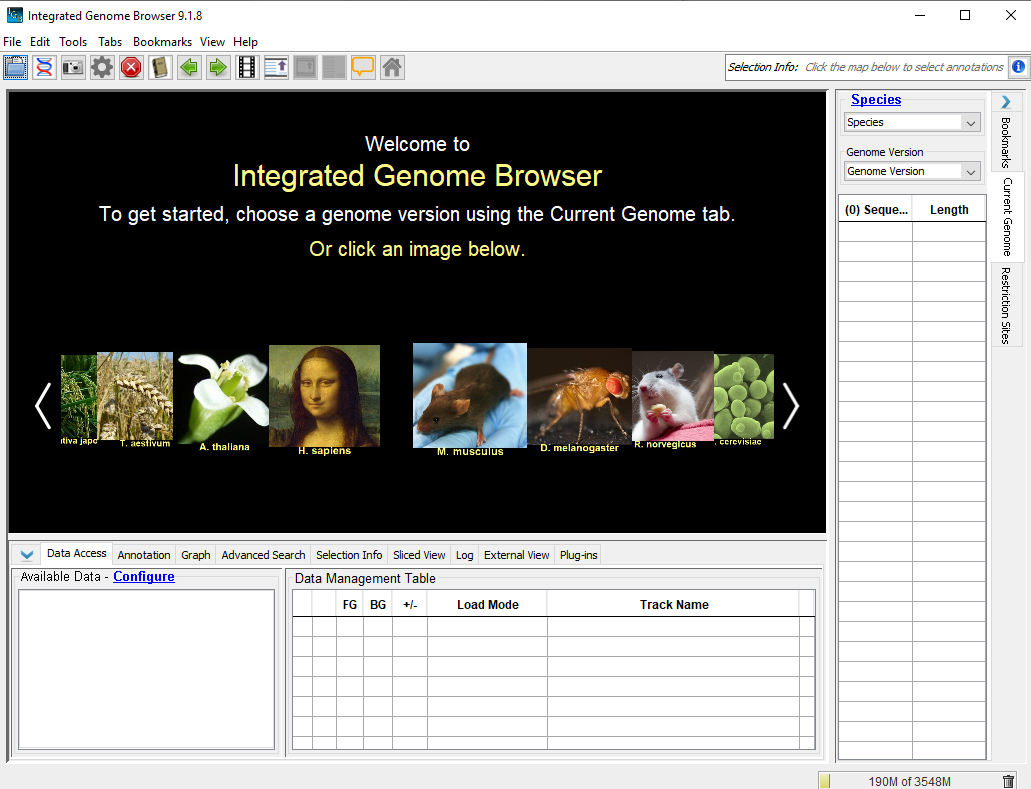 * Open the fasta sequence as a genome
* Open the fasta sequence as a genome
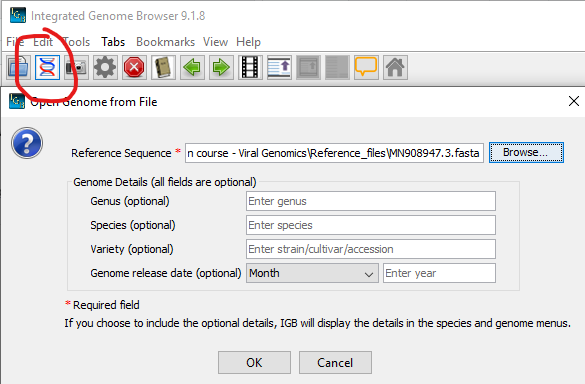 Remember to ’Load Genome when are back on main screen
Remember to ’Load Genome when are back on main screen
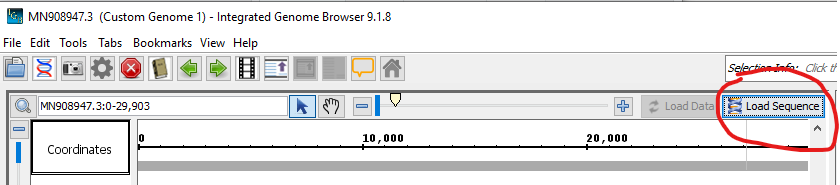
- Add and load gff
open gff > File > Open File > [Select gff from file browser]
Load data
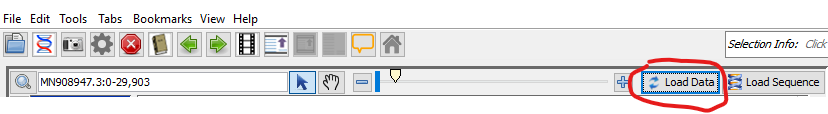 You should now see a preview of the annotated genome looking something like this:
You should now see a preview of the annotated genome looking something like this:
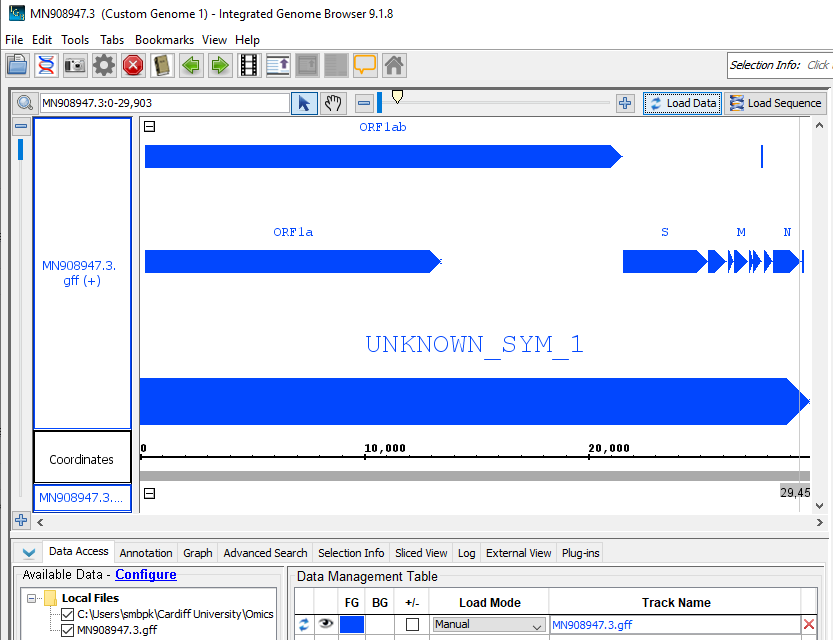
- Add mapped primertrimmed sorted bam
File location:
Omics revolution course - Viral\ Genomics\artic_analysis\ncovIllumina_sequenceAnalysis_trimPrimerSequences\SRR13018901.mapped.primertrimmed.sorted.bamopen bam > File > Open File > [Select bam from file browser]
Now load data as you did previously.
You should now see a preview of the annotated genome with bam overlay looking something like this (it may take time to appear):

use slider and top to zoom in to see the primer masking and SNPs:
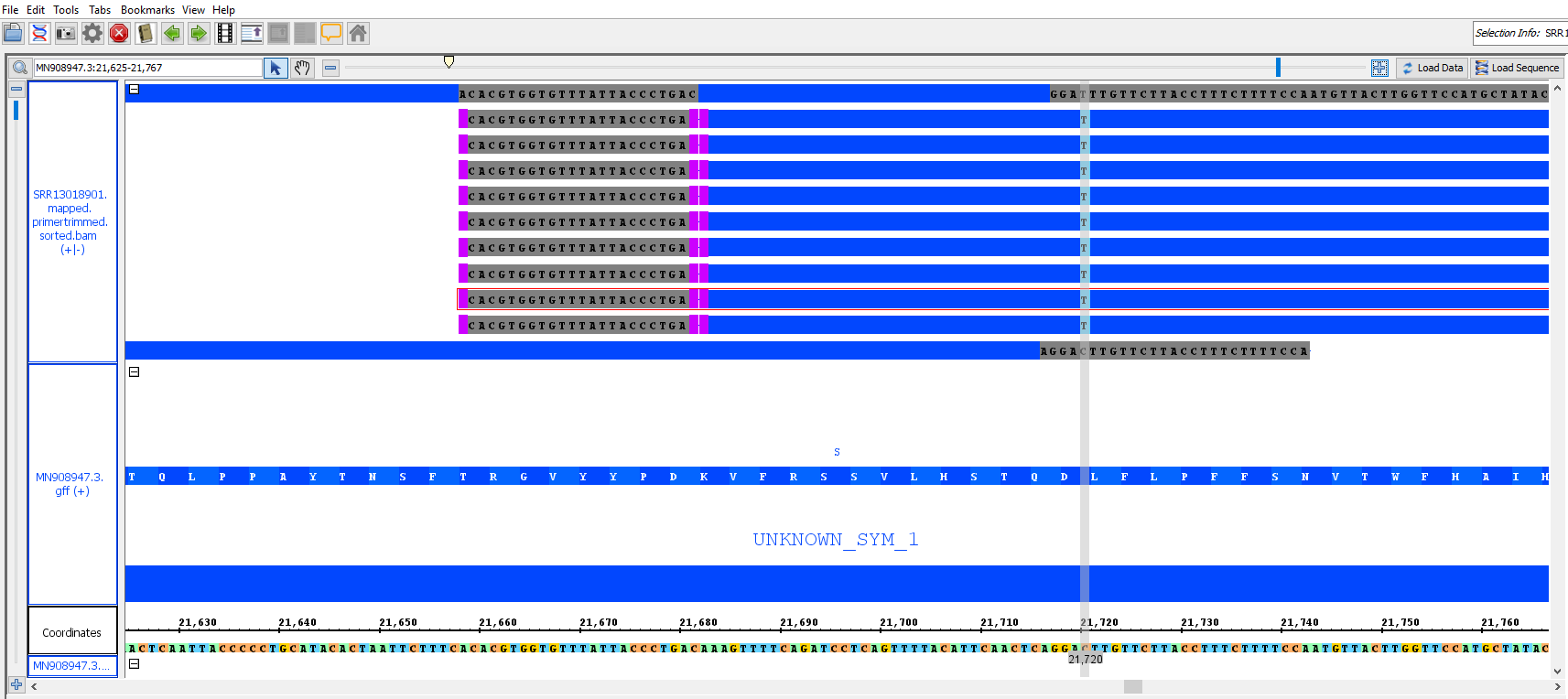
- Explore settings on IGB to visualize the primer sites and mutations - identify synonymous and non-synonymous mutations
Explore the Varient amino acids encoded by the sequence (this file can be opened in Excel)
Omics revolution course - Viral Genomics\artic_analysis\artic_test_run_221022.variant_summary.csv
| sampleID | gene | aa_var | dna_var |
|---|---|---|---|
| SRR13018901 | ORF1ab | Syn.924F | C3037T |
| SRR13018901 | ORF1ab | Syn.1749N | C5512T |
| SRR13018901 | S | Syn.53D | C21721T |
Phylogenetic Classification of Concensus Viral Sequnece
Data
Data was download from GISAID representing viral sequences from across (time and space) of the pandemic.
These sequences can be found in: Omics revolution course - Viral Genomics\Viral_sequences\SARAS-CoV-2
EPI_ISL_14569384.fasta
EPI_ISL_14871243.fasta
EPI_ISL_15038461.fasta
EPI_ISL_15165123.fasta
EPI_ISL_15196344.fasta
EPI_ISL_15233297.fasta
EPI_ISL_15251359.fasta
EPI_ISL_15266729.fasta
EPI_ISL_15266749.fasta
EPI_ISL_15283205.fasta
EPI_ISL_15395946.fasta
EPI_ISL_15400456.fasta
EPI_ISL_15422334.fasta
EPI_ISL_15424763.fasta
EPI_ISL_15426260.fasta
EPI_ISL_15436037.fasta
EPI_ISL_15458335.fasta
EPI_ISL_15463432.fasta
EPI_ISL_15466700.fasta
EPI_ISL_402128.fasta
EPI_ISL_529216.fastaSoftware
Nextclade is a tool that identifies differences between your sequences and a reference sequence, uses these differences to assign your sequences to clades, and reports potential sequence quality issues in your data. You can use the tool to analyze sequences before you upload them to a database, or if you want to assign Nextstrain clades to a set of sequences.
To analyze your data, drag a fasta file onto the upload box or paste sequences into the text box. These sequences will then be analyzed in your browser - data never leave your computer. Since your computer is doing the work rather than a server, it is advisable to analyze at most a few hundred sequences at a time.
The source code for the application and algorithms is opensource and is available on GitHub. User manual is available at docs.nextstrain.org/projects/nextclade.
Workshop exercise
- Load run sequence analysis
- Review the Results

Click / hover over the element of the analysis to see more detail
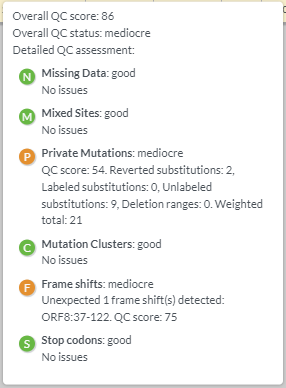
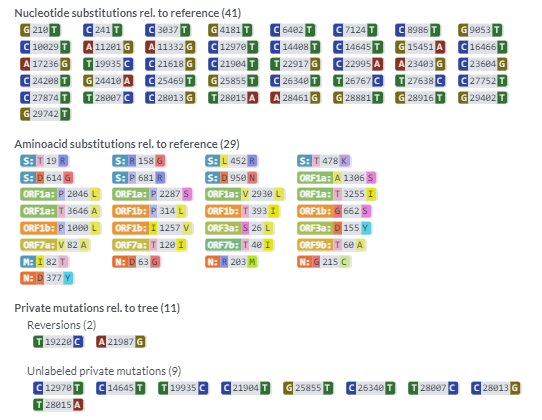
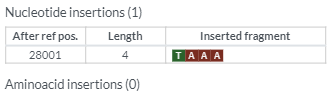
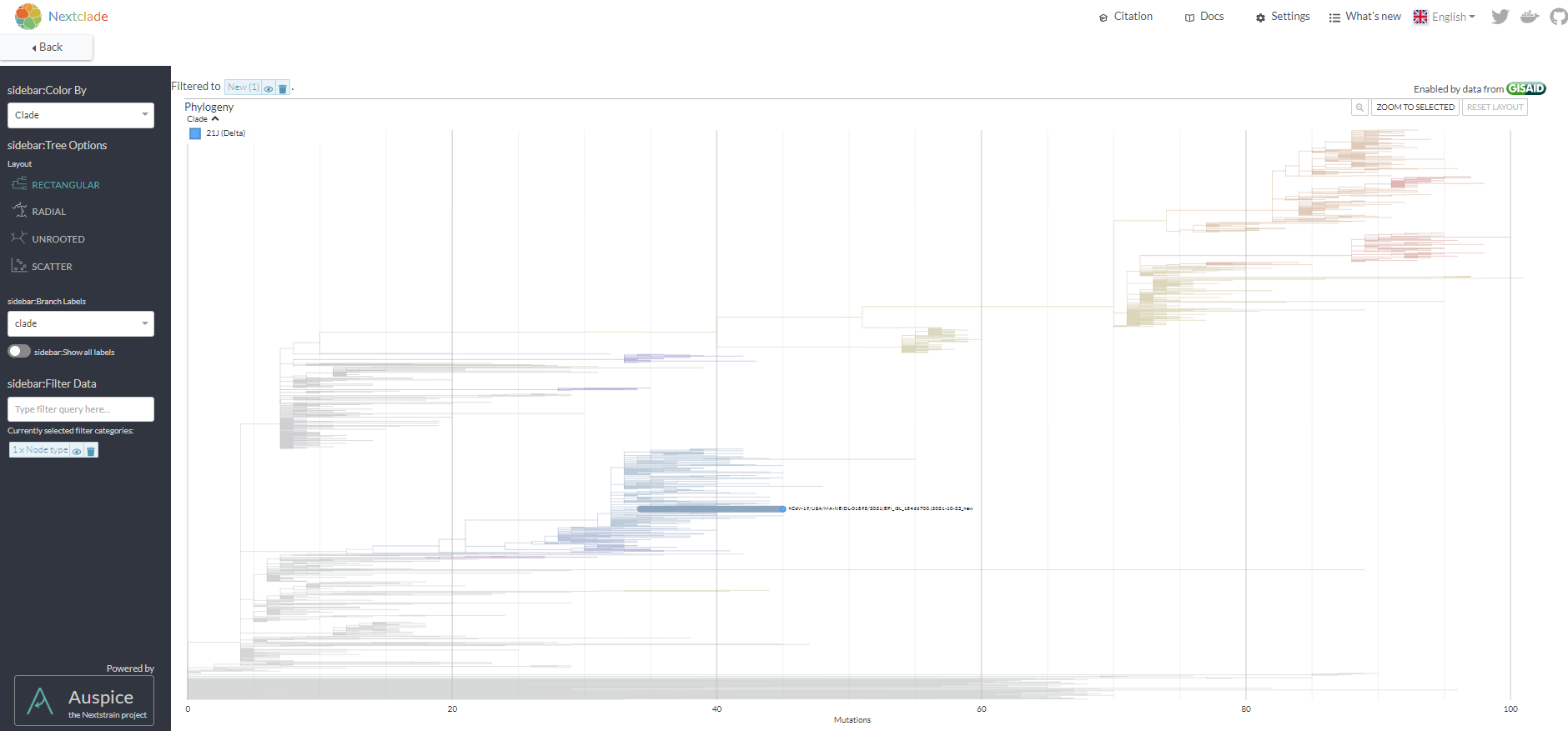
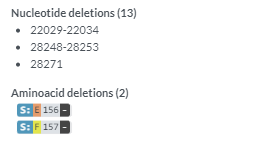 * 3. Review the position fall on the phylogenetic tree
* 3. Review the position fall on the phylogenetic tree
Click on the tree icon at the top right of the screen
- Download and review results
Click on the download icon at the top right of the screen
 I suggest using nextclade.tsv and opening file in excel.
I suggest using nextclade.tsv and opening file in excel.
- Now repeat the analysis with other sequences provided
- Can you determine where and when each sequence was derived from ?
Structural and Varient analysis
Data
Data was download from GISAID representing viral sequences from across (time and space) of the pandemic.
These sequences can be found in: Omics revolution course - Viral Genomics\Viral_sequences\SARAS-CoV-2
Software
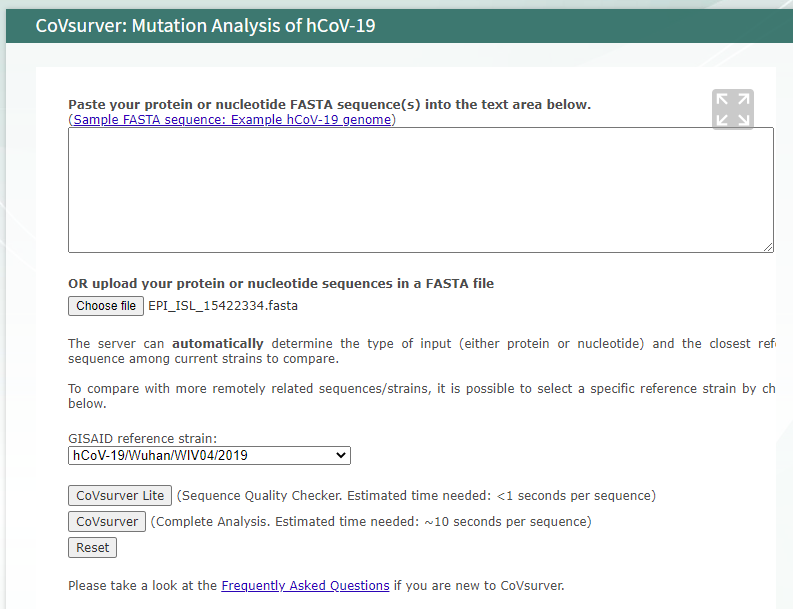
Workshop exercise
- Upload and analysis sequences provide
You should see the following outputs - discuss how this help in fighting the pandemic ?

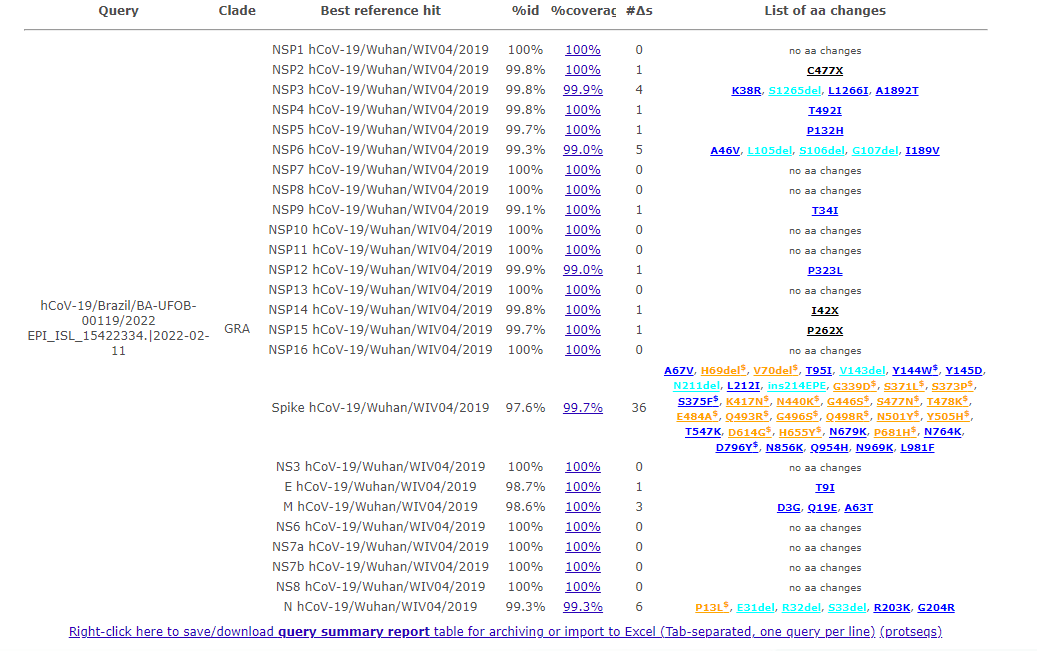
Covsurver_varients